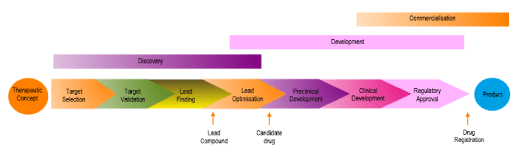
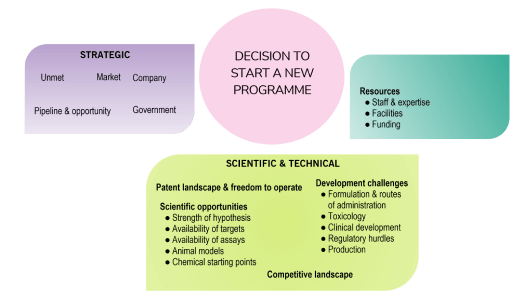
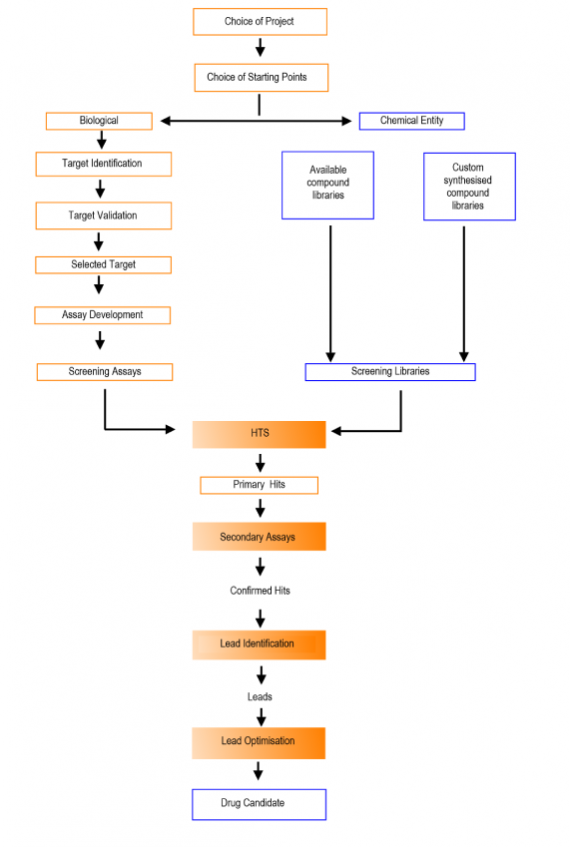
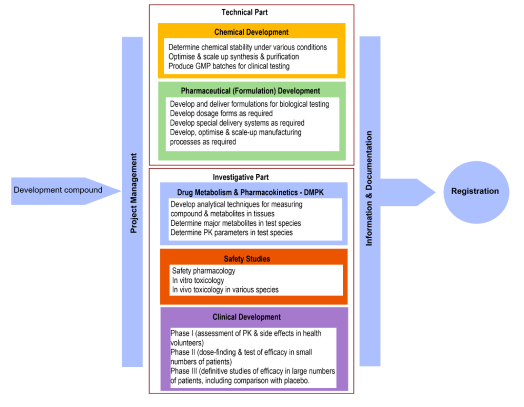
Here are 6 tips to effective presentations
Better than average communicators are generally more successful than most people, but great communicators are the ones that start movements. They are remembered long after their speeches. Think Jefferson, Churchill, Gandhi, Kennedy, King, Mandela and Obama, to name but a few.
Failure to communicate effectively in science can means research won’t get funded, products won’t get sold, projects won’t get backing, and careers won’t thrive. As career scientists, your ability to deliver captivating talks can mean the difference between acclaim and toiling in obscurity.
So here are my six simple tips you can adopt in 2022 to move your presentations to a new level, enabling you to communicate in ways that are passionate, powerful, and inspiring.
Tip #1: Unleash the Master Within
Thomas Jefferson, the third President and Founding Father of the United States of America is highly regarded even today as one of America’s most influential leaders. He was a passionate believer in democracy and considered it essential to the expression of society. He promoted national self-determination, public education, and a free press.
As the principal author of the United States Declaration of Independence, he wrote many inspiring speeches, which went on to shape the course of history. The preamble to the Declaration of Independence, for example, evokes the original spirit of the American nation:
‘We hold these truths to be self-evident, that all men are created equal, that they are endowed, by their Creator, with certain unalienable Rights, that among these are Life, Liberty, and the pursuit of Happiness….’
Passion and public speaking are intimately linked. To touch your audience, you need to dig deep to identify how you’re uniquely and meaningfully connected to your presentation topic. This is where you’re operating at people’s emotional level. Passion is your why, or inspiration. It is not a passing interest or a hobby but rather that thing that’s core to what makes you, you! It is what gives you the authority, mastery and command, and your presentation will be empty without it.
Bear in mind that in some situations, what fires you up might not be obvious. Often, it is dressed up as something else. Howard Schultz, the former Chairman and Executive of Starbucks once said his passion was not coffee, but rather creating a third place between work and home! Coffee was only the by-product.
Passion is what makes successful speakers always enthusiastic about sharing their ideas. They have bags of charisma. They radiate joy and positivity about their ideas, and they are motivated by ‘good’ intentions, such as a desire to make a difference, create impact or leave a legacy.
Just as we know that happiness at and passion about the work are vital to career success, it is the same with public speaking. If you’re not having a great time in your job, how do you expect to generate enthusiasm in your presentation about it?
So while we can talk about effective storytelling, designing beautiful PowerPoint slides or how to use body language more effectively in your public speaking but the fact, and it is a fundamental fact, that effective presentations require passion first. Effective stories, slides or body language mean little if the speaker does not radiate passion and enthusiasm about what they’re communicating.
Tip #2: Master the Art of Storytelling
In this information-saturated age that we live, you won’t be won’t be heard unless you tell compelling stories. Facts and figures, and all the rational things that we think are important in science actually don’t stick in our minds that well. However, stories create “sticky” memories by attaching emotions to things that happen.
Stories also affirm who we are. We all want affirmations that our lives have meaning. And nothing does a greater affirmation than when we connect through stories.
This is why people who know how to weave stories about their work and share good stories have a powerful advantage over others.
But what constitutes a good story? Consider the case of major film studios, such as MGM, Pixar and Disney. They have individually mastered the ability to move audiences deeply, causing adults to tear up next to children, while persuasively transporting us into make-believe worlds.
Their perennial success in the business of movies is down to the way they choose ideas, create compelling characters, invoke empathy, drama and conflict, create villains and heroes, and the endings (the moral), that is, storytelling. It is the same with great speakers.
Aristotle, the Greek philosopher, believed that persuasion happened when three components were represented: ethos, logos, and pathos. Ethos is credibility. We tend to trust and agree with people we respect for their achievements, titles, experiences, etc. Logos is about persuasion through logic and data. Pathos is the act of appealing to emotions.
You can see this approach in Stevenson’s TED talk. For instance, he started with his personal experiences. The first five minutes (30 percent of the presentation) were on his personal stories and experiences. Data about incarceration in U.S. prisons came in later to support his ideas. He chose his approach to make it easy for the audience to connect with him on a personal and emotional level.
Studies have shown that inspiring communicators use three types of story.
The first types of story are personal stories about who we are. They should be descriptive and rich with imagery to enable the listener to imagine themselves with you at the same time. Delivered well, a captivating story makes your audience know something about you, which builds trust. Granted, personal stories are a sensitive subject, but if you choose them carefully, nothing comes close to grabbing the audience’s attention early on. A personal experience that produced an unexpected outcome often works well. The key thing is not to make them show how great you are, etc.
The second types of story are stories about other people who have learned a lesson the audience can relate to. The power of such stories is that they shed light on our shared humanity. So while personal stories can evoke empathy, it is stories about other people that audiences mostly empathise with. Empathy is the capacity to recognise and feel others’ experiences.
The third type of story are stories about successes or failures of products or brands. Harvard Business School is famed for the Case Method to teaching MBA students. These cases usually tell stories (real or simulated) about challenges faced by business executives and lessons that can be learnt from their experiences. This way, students are able to relate to business theorems with particular challenges.
Just as a great novel or movie goes about storytelling, a great presentation has to have a narrative, a cast of characters (hero and villain) and the moral of the story. The story should reveal a challenge (villain) being faced, a protagonist or hero (your solution) who is committed to rising to the challenge, the townspeople (customers) to be freed by the villain, and the outcome (the people who will be freed and live happily ever after their struggles are ended).
Tip 3: Have a Conversation
Great speakers deliver their content in a natural, authentic way, akin to having a comfortable conversation with a friend. It is a skill learned through practice and is not something that can just be memorised and perfected in an instant.
Think of the times you had a genuine conversation with a friend. Hopefully, you’re typically operating in a zone of emotional rapport. You were able to persuade your friend because you had gained their trust, and your voice, gestures, and body language were all in sync with your words.
This authenticity does not happen spontaneously. It is something that is learned, through practice. It takes hours of practice, searching for the right words that best represent the way you feel, delivering those words in a powerful way for maximum impact.
Good verbal delivery is based on what is called in the military as ‘commanding presence’. Commanding encompasses the following key elements:
- Rate: the speed at which you speak
- Volume: the loudness or softness
- Pitch: high or low inflections
- Pauses: short pauses to put emphasis on key words
- Gestures, facial expressions, and body language
Great communicators speak at the right rate (the ideal rate of speech is between 180 and 200 words per minute), they speak concisely and precisely, and their voices project across the entire room because they speak from their diaphragms. They compliment the words with the gestures and facial expressions, to make a strong argument even stronger.
Tip 4: Reveal Something New
Great speakers incorporate new information or perspectives that are completely new to their audiences. The information may be packaged differently or presented in a way to solve an old problem. Revealing new perspectives works because our human brains love novelty. Unfamiliar, unexpected or unusual outcomes in a presentation audience, jolts them out of their preconceived notions, and provides them with new perspectives.
One of the most captivating public speakers on the web today is Professor Hans Rosling. He often talks about population, economic development and global health issues. As well as delivering data in a fascinating and easy-to-digest way, he is able to reveal completely new perspectives.
This is the same approach taken by all successful communicators. They opt to deliver content in ways that reveal something that is entirely new; things the audience was not familiar with.
Seth Godin, the popular blogger and author, has made a career out of delivering ideas differently. He told a TED audience in 2003 that in a society with information overload, the natural instinct is for audiences to ignore most of it. Thus, delivering the same old, tired content using the same boring methods as everyone else is bound to fail. Adding a little spin to content allows the audience to be more receptive to the message.
Tip #5: Incorporate Jaw Dropping Moments
A jaw-dropping moment in a presentation is when the speaker delivers a shocking, impressive or surprising moment that is very moving and memorable that it grabs the audience’s attention, and is remembered long after the presentation is over. Jaw-dropping moments are capable of heightening emotions, helping listeners recall and act on the message.
In 2009, Bill Gates, the founder of Microsoft delivered a talk at a technology conference about malaria. While on stage, he opened up a glass jar and said, “Malaria is spread by mosquitoes. I brought some here, just so you could experience this. We’ll let those roam around the auditorium little bit. There’s no reason only poor people should have the experience.” The audience roared with laughter, cheered, and applauded. Bill Gates had effectively delivered his jaw-dropping moment.
A few sentences earlier, Bill gates had talked about how many children lives’ could be saved through better medicines and vaccines. He was able to deliver an emphatic talk. He used shock and humour to drive his point home.
Journalists call the mosquito gimmick “the hook.” It’s the wow moment, the showstopper and the device used to capture the audience’s attention. Used cleverly, it allows listeners to share your story. So, before creating a Power Point presentation, take time to think about the story first. In the same manner a movie director storyboards the scenes before shooting, you should create the story before opening the tool. Aim to tap into al the senses – seeing, touching, feeling, and smelling.
Things that shock, surprise, bring fear, joy or wonder impact how vividly we remember them. It is the reason many of us remember our first kiss, the birth of a child, winning an award, break-ups or death of a loved one. It is as though these emotionally charged events are burned into our memories. Therefore, if you want to connect with an audience in an emotional level, you will need to present information that is vivid, using tools and examples that meaningful and concrete.
Tip #6: Be mindful of Cognitive Backlog
Most memorable presentations are noted for three key elements:
- Are concise and organised systematically
- Use multisensory approaches to paint mental pictures in their audiences
- Are authentic, open and transparent.
Conciseness and Organisation
It is an undeniable fact that listening is mentally draining. Thinking, speaking and listening are physically exhausting. Think of the last time you sat through a one-hour lecture or power Point presentation. Too much information prevents the effective transfer of ideas, leaves the audience anxious and even frustrated. Researchers refer to this information overload as “cognitive backlog,” which is akin to piling on weights, which makes the mental load heavier.
This is the reason all TED talks are required to be no more than 20 minutes. TED believes that 20 minutes is short enough to hold one’s attention, and long enough to cover anything relevant.
If you must give longer presentations, it is necessary to split them into chunks, for instance, by adding breaks, videos, stories or demonstrations, every 10 minutes. The longer the presentation, the more the listener has to work to organise, comprehend and recall information.
John F Kennedy, the 35th president of the United States, gave a famous speech at Rice University in late 1962. It was here that Kennedy outlined his vision for America to explore the moon. The speech, which lasted just over 17 minutes, captured the nation’s imagination about the importance of exploring space.
But it is not enough to be concise. In fact conciseness means nothing if the information is haphazard and unstructured. This is why some influential communications professionals talk of the rule of threes. This rule simply means that people remember three pieces of information well. Add more items and retention starts to wane quickly.
To make use of the rule of three, structure your story in three key chunks or messages around a central theme. It turns out that the rule of three pervades our work and social lives on a daily basis. You will find it in literature (the three little pigs and, the three musketeers), in the arts (three primary colours), politics (the three arms of government), etcetera. If it works for the world’s greatest writers and painters, it will work for presentations, too.
Use of Multisensory Experiences to Paint Mental Pictures
Think again about a particularly boring talk you had the misfortune of attending. What made it boring? What was your level of engagement? Chances are that it had too much text, lacked structure, was visually unappealing and the content was unengaging.
The fact is that boring does not wash well with the human brain. The brain craves multisensory experiences and will quickly switch off when it is exposed to stuff that is boring. Having presentations that include more than one sense: sight, sound, touch, and smell are difficult to ignore. This is why great talks use mesmerizing images, captivating videos, intriguing props, beautiful words, and more than one voice to bring the story to life.
Granted, some of these experiences, such as smell and taste, are difficult to incorporate in presentations. The key thing is to build a presentation around one or two main senses, and incorporate one other. The harder experiences can be simply described.
Slides should incorporate images and videos rather than text whenever possible. The audience is far more likely to recall information when it is presented in a combination of pictures and text rather than text alone.
The other important sense to use is sound. The auditory sensation is very powerful and how the content is delivered (pitch, rate, volume, intensity, sound effects) can all touch the listeners soul.
The final sensation to use is feeling. Feeling has been described as the “holy grail” of presentations owing to its ability to transport audiences to another place. The visual display of information helps the audience to see it while touching allows them to complete the journey.
Being Authentic, Open and Transparent
Although public speaking is an artform, it is not act one can put on. Am sure you have met a person who acts and speaks one way in private only to sound completely different when delivering a presentation. Such people act, look and sound like two different people. They lack authenticity, openness and transparency. Unfortunately, audiences are not thick – they can see through a fib, so trying to be somebody you’re not is a sure way to fail at building rapport with your audience.
If your goal is to inspire the audience and take them with you, you must be real. Here are some things to do:
- Use your own voice – there’s no need to sound ‘posh’ or adopt some ‘esoteric voice.’ Chances are that it will make it difficult for your audience to keep up.
- Disregard the fact this is a presentation. Instead, regard it as a conversation, the kind you typically have with family and friends.
- Relax! This is not a sermon on the mountain, rather you’re just sharing your knowledge and expertise for people to take as much, or as little, as they wish.
- Be yourself – you’re fantastic at it!
Finally, try to recapture your inner 3-year old-the times you were carefree, and had no hang ups. If you can get back to that, you’ll be an impactful public speaker.