Since the thalidomide disaster, ensuring that medicines are safe before they are approved for sale to the public has become one of the most important tasks of regulatory agencies.
However, while most people are familiar with clinical trials and how they are used to assess safety and efficacy of new medicines (technically, the active ingredients), excipient safety is rarely featured, and has remained a mystery.
In this short article, we will explain how the safety of different pharmaceutical excipients, such as parabens and vaccine adjuvants, is assessed by pharmaceutical scientists today.
What is excipient safety?
Whether a particular excipient is safe or not depends on what safety actually means. The commonly agreed definition of safety is the absence of undesirable pharmacodynamics effects (adverse and side effects) that a particular substance exerts on the physiological function of the living organism upon exposure within its therapeutic range.
Excipient toxicity, another closely related term to safety, and often used interchangeably, actually refers to the degree a material is poisonous, i.e its ability to cause injury.
Undesirable effects include sensitization, allergic reactions, anaemia, jaundice, kidney damage, nerve damage, interference with vision, blood cell count, et cetera.
It is important to also note that safety and toxicity depend on many other factors, not just the dose, duration or route of exposure of that material, per se. This is sometimes referred to as the intrinsic safety/toxicity.
Some excipients subtly alter physiological properties, such as increased gastrointestinal motility, permeability of membranes, metabolism and excretion. This type of toxicity is known as extrinsic.
Examples of undesirable effects in commonly used excipients are shown in the table below.
| Excipient name | Function in the product | Adverse effects |
|---|---|---|
| Propylene glycol | Solvent | CNS effects, especially in children under 4 years old |
| Benzyl alcohol | Preservative | Hypersensitivity reactions, respiratory system effects |
| Ethanol (alcohol) | Solvent and co-solvent | Intoxication |
| Polysorbates | Solubilising agent & surfactant | E-Ferol syndrome |
| Hydrogenated castor oil derivatives | Vehicle and solubilising agent | Anaphylaxis |
| Parabens | Preservative | Reports of oestrogenic effects, especially for propylparaben. Also hypersensitivity reactions & anaemia in neonate |
| Saccharin | Artificial sweetener | Hypersensitivity and photosentivity reactions |
| Glucose and sucrose | Sweetener | Dental caries |
| Benzalkonium chloride | Preservative & surface active agent | Bronchospasm when used in combination with asthma drugs. Ear damage |
| Sodium metabisulfite | Antioxidant | Wheezing and chest tightness in asthmatic children |
| Aspartame | Artificial sweetener | Contra-indicated in patients with phenylketonuria |
| Colours (e.g Azo dyes) | Colorants | Sensitivity reactions. Some reports of hyperactvity in children |
| Chlorhexidine (all salts) | Biocide & preservative | Hypersensitivity reactions, irritation of conjuctiva and corneal damage in high concentrations |
| Menthol | Flavour | Inhalation in large quantities causes ataxia and CNS depression. Hypersensitivity reactions are reported |
| Lactose | Filler and diluent | In lactose intolerant individuals, cramps, diarrhoea and flatulence |
Safety of an excipient is formally assessed by monitoring its effects on vital systems, in particular, the cardiovascular, the central nervous and respiratory systems.
A material’s toxicity will depend on a variety of factors, including the dose of the substance, duration and route of exposure.
For a long time, excipients were considered inert substances, included in products to facilitate production and/or improve convenience during use.
This definition, in the strictest sense, now only applies to natural, simple substances, such as honey, simple sugars and other substances that are well characterised and known to be inert.
The importance of safety
Modern pharmaceutical products are anything but simple mixtures; they are complex engineered products in which several ingredients are combined to produce specific characteristics.
Many of these complex materials have the potential to adversely affect consumers, particularly if used in large quantities.
For this reason, there is a process for ensuring that the safety of every ingredient in the formulation is well studied and mapped out.
Safety is especially important since excipients are often present in very high proportions relative to the active ingredient.
But safety is also dependent on the way a product is handled, for instance during formulation or manufacturing, it may be exposed to high heat or humidity, leading to an increase in the probability to generating secondary products and impurities to be introduced into the product.
During use, the patient may not store is correctly, leading to further degradation to produce toxic impurities.
Excipients may also interact with the medicinal agent, other excipients in the formulation, the container or the atmosphere, leading to many more undesirable effects.
Thus, safety and toxicity of the pure substance as well as any degradants must be known, both by regulatory agencies and product manufacturers, and rightly so, by the public who are the ultimate users of medical products.
Methods used to assess excipient safety
Mapping out the intrinsic and extrinsic safety, as well as understanding potential risks and post-approval monitoring are what constitute the universe of excipient safety assessment.
 Some of the tests and investigations conducted on excipients to understand their instrinsic and extrinsic toxicological profiles are shown in the infographic below.
Some of the tests and investigations conducted on excipients to understand their instrinsic and extrinsic toxicological profiles are shown in the infographic below.
Note that many of these resemble those tests done on new chemical entities: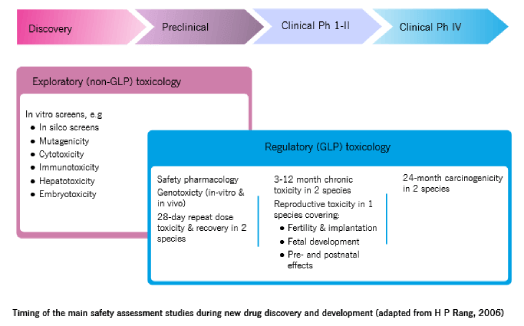 The challenge for this approach, however, is that there can be no end to it – rather like the proverbial length of a piece of string.
The challenge for this approach, however, is that there can be no end to it – rather like the proverbial length of a piece of string.
This problem (of unending safety assessment) can be overcome by adopting a risk-based view of safety; by seeking to understand and address any potential risks during the life history of the material, from the earliest stages of synthesis, through to product development, launch and use.
Risk-based assessment of safety
A risk-based approach to safety and toxicity requires manufacturers of the excipient and medical products to adhere to good manufacturing practices (GMPs) when handling excipients, very much similar to those for active ingredients and follow guidelines set by regulatory agencies.
Some of the requirements from regulatory agencies include routine testing of excipients to ascertain and verify a material’s identity, purity, traceability of batches, and quality, which are all important determinants for safety and efficacy of the product.
The particular safety assessment pathway taken is largely dictated by precedence of use. The exact requirements depend on the regulatory agencies, which operate under different rules.
For practical reasons, we’ll focus on three regulatory agencies: the US Food and Drug Administration (FDA), the European Medicines Agency, and the Japanese Ministry of Health, Labour, and Welfare:
Excipients with precedence of use
If an excipient has already been used in an approved pharmaceutical product, is a food additive, or any product with human exposure, that is, where previously safety tests were carried out, it is considered to have precedent.
Such materials are already registered for use and may be included in official pharmacopoeia or compendia.
The US Food and Drug Administration (FDA) has an excipient database (FDA Inactive Ingredients Database) which lists all excipients and their precedence of use.
There is also the United States Pharmacopoeia and National Formulary that carries monographs of approved excipients for use there. You can click here to access the Inactive Ingredients Database.
Related post:
- Full list of excipients Generally Recognised As Safe
The European Medicines Agency does not have a list of ingredients; however, individual member countries have compendia that list approved materials and their routes of administration.
In France, for instance, this is the “Dictionnaire Vidal,” and in Germany the “Die Rote Liste”. The European Pharmacopoeia, just like the USP-NF carries monographs of excipients approved across the bloc.
In Japan, the Japanese Ministry of Health, Labour, and Welfare has a precedence of use compendium known as the Japanese Pharmaceutical Excipients dictionary, and the Japanese Pharmacopoeia. Both show the different excipients that have been previously used in drug formulations in that country, with the description of their characteristics, and the maximum dose used in the different administration routes.
Excipients with no precedence of use
Excipients that have never been used in any products and therefore have no safety assessments undertaken formally are considered novel excipients.
Novel excipients are materials being used in a drug product for the first time or a new route of administration.
In other words, they are not included in the Inactive Ingredient Database, United States Pharmacopeia-National Formulary (USP-NF), European Pharmacopoeia, Japanese Pharmacopoeia, or other compendia, including the “Handbook of Pharmaceutical Excipients.
Also, approved excipients that have undergone chemical modification, however minor, are considered novel excipient by regulatory agencies.
For these materials, there are several guidelines in operation by respective regulatory authorities before they can be permitted for use in medicines.
For instance, the US FDA requires nonclinical and clinical assessments to be undertaken. These studies not only aims to assess the excipient’s intrinsic safety but also the extrinsic Safety profile.
For further reference, please see USP-NF 26 General Chapter <1074 > “Excipient Biological Safety Evaluation Guidelines”, and IPEC’s New Excipient Evaluation Guidelines.
The excipient manufacturer is expected to develop full safety information recommended in the guidelines, based on the material’s future use and dosage form.
The battery of tests to be conducted are described in these guidelines.
Excipient Safety Assessment: Two common Questions
Pharmacopoeia: Are excipients required to be listed in the pharmacopoeia?
A common question that often arises is whether an excipient is required to have a monograph before it can be commercialised. The simple answer is no. There is no regulatory requirement for a monograph.
However, it is always preferable to present an excipient with a monograph or a complementary document that is relevant, because it means that such a material has established quality specifications for pharmaceutical use.
If the excipient is present in several pharmacopoeias, the manufacturer is expected to ensure conformity for all monograph requirements, present in all pharmacopoeias, before its commercialization to the global market.
The manufacturer demonstrates that methods used to test the material are valid and provide assurance that the excipient, when tested by these methods, conforms to all requirements.
When the excipient is not present in any pharmacopoeia or other monograph in a compendium, the manufacturer can establish an own specification, based on a similar monograph present in the pharmacopoeia.
Pharmacopoeia: Can excipients with no monographs be used in medicines?
An excipient that is not written in any pharmacopoeia can be used in a drug product, even if there is no monograph. However, regulatory authorities normally require a detailed evaluation of the excipient’s safety and toxicity, which can be an expensive endeavour.
If there is no monograph available, a specification can be based on the manufacturer’s own experience and in the chemical and physical properties of the excipient according to the intended use of administration.
Inclusion of a novel material into the pharmacopoeia depends on its commercial importance and its use in a pre-approved drug product.
In this case, both the excipient manufacturer and medicinal product manufacturer can request regulatory agencies or the national pharmacopoeia commission to draw up a new monograph, provided the novel excipient has received approval through its use in a marketed drug product.
At this stage, the novel excipient is considered approved for use by the stated route of administration and in the stipulated dosage levels as in the approved drug.
What about Drug Master Files?
The first step to introduce a new excipient in the market is the determination of its functionality, route of administration, identification, and stability of the intended API for a drug formulation.
Once these steps are all set, a document with all information known as a Drug Master File (DMF), or European Certificates of Suitability (CEP) is created.
This document provides all the technical information such as specifications and test methods for raw materials, in-process testing, the finished excipient product, manufacturing process, safety, packaging details, and content label.
In the United States, the supplier submits the DMF to FDA, which is held in confidence by the administration. In Europe, the DMF only exists for APIs and is not available for excipients.
CEPs are used for pharmaceutical excipients, which may be treated in two ways: CEPs for excipients listed in Ph. Eur and CEPs for excipients not listed in the Ph. Eur.
Other supplementary tests for assessing safety
Excipient assessments for generic drug products
During generic product development, the sponsoring company may choose to use excipients that are different from the innovator formulation either because of a change in route of administration or dosage forms.
In this case, the formulations and or route of administration are different but the API is already established and known.
In both cases, excipients will be selected based on the function they perform, compatibility with the API and other formulation components and any other considerations, such as cost, material availability and the company’s own experience and preferences.
Regulatory approval will only be granted if it’s established that the excipients are suitable to be used in the new formulation, via the intended route of administration or the dosage form specified for the new drug product.
Investigations of Excipient – API Incompatibilities
Since some excipients react with and can modify excipients or the API, additional compatibility studies between excipients and APIs are mandated. Such reaction products may be harmful or alter the stability of the API.
There are many ways excipient compatibility studies can be done, the most common ones being differential scanning calorimetry (DSC) or isothermal calorimetry, using simple blends of the excipient and API in simple ratio.
Thus, assessing the nature of these interactions therefore aims to select only materials that are compatible with the drug and will not compromise the stability or safety of the formulation. If incompatibilities are established, the formulation team will aim to alleviate the incompatibilities.
Presence of impurities and residual solvents
The presence of impurities and solvents in the product formulations need to be identified and controlled within specified limits. Impurities and solvents can be introduced during the material’s manufacture, product formulation or production.
Residual solvents are classified into three main groups, depending on their toxicity and potential harm:
Class 1 solvents are to be avoided in the formulation because they are known or suspected carcinogens and hazardous to the environment. Examples of Class 1 solvents include carbon tetrachloride and benzene.
Class 2 solvents are to be limited because while being non-genotoxic are suspected to have neurotoxicity or teratogenicity, or suspected to be causative agents of the same. Examples of Class 2 solvents include acetonitrile, methanol and dioxane.
Class 3 solvents have low toxic potential to humans however, they may be limited in terms of the total quantities allowed in the formulation. Examples of Class 3 solvents include ethanol, acetone and ethyl acetate.
Excipient stability studies
Excipient stability studies are carried out with the aim of determining whether the material maintains its quality over the time period it is used. The excipient will be studied under various environmental conditions, such as high temperature, light and humidity.
A major objective of excipient stability studies is to determine the expiry date or re-test period, which refers to the time period within which the raw material is expected to remain within specification, and therefore, safe for use in the drug product. A material that has expired and or has not been re-tested is not safe for use, and should be destroyed.
Market surveillance
The final, and by no means the least aspect of safety assessment is market surveillance post-launch of the product onto the market, whereby healthcare professionals and the public report any side and adverse effects encountered into a central database. The data are then collated and analysed for patterns and trends that might point to previously unreported toxicity to ensure the product is withdrawn before it can harm more people.
The significance of market surveillance is evident when you consider the fact that twenty percent of adverse effects and toxicities are discovered during this phase of market surveillance.
Conclusions on excipient safety
For a long time only the quality, efficacy, and safety of the API was considered as the most important aspects of a new medicine.
This approach has now changed, and manufacturers are required not only to study an excipient’s safety but they must also justify its inclusion in a product.
Any ingredient used in the product must be beneficial and help optimize the performance of the product, both during the manufacture and also during use.
The process of assessing safety involves a battery of established tests that determine the physiological effects of the material, including potential effect on the cardiovascular, respiratory and central nervous system.
In addition, any interactions with other components in the product must be studied and understood fully.
The rule of the thumb is to simplify formulations as much as possible and reduce the number of excipients to those that are absolutely necessary.
Sources used
American Academy for Paediatrics: Committee on Drugs. ‘Inactive’ ingredients in pharmaceutical products update (Subject review). Pediatrics 1997;99(2):268–278. Available at: http://pediatrics.aappublications.org/content/99/2/268.long (accessed August 2021)
Turner MA, Duncan JC, Shah U et al. Risk assessment of neonatal excipient exposure: lessons from food safety and other areas. Adv Drug Deliv Rev 2014;73: 89–101. DOI:10.1016/j.addr.2013.11.003
C.L. Winek, History of excipient safety and toxicity, Drugs and the pharmaceutical sciences, 103 (2000) 59-72. PMID: 27262205. DOI: 10.1016/j.xphs.2016.03.019